ADVERTISEMENT – THE CONTENT OF THIS ARTICLE DOES NOT FALL UNDER THE RESPONSIBILITY OF THE EDITORIAL BOARD. ARTICLE WRITTEN ON REQUEST OF PFIZER.
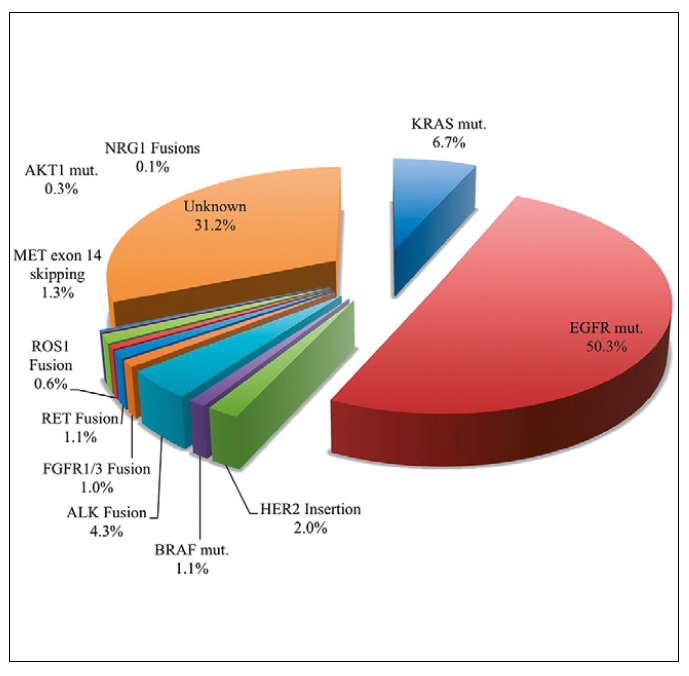
The identification of recurrent gene rearrangements involving the anaplastic lymphoma kinase gene (ALK) in a subgroup of patients with non-small cell lung cancer (NSCLC) was an important milestone in the shift towards a more personalized treatment strategy for this disease entity.1 ALK rearrangements are found in 3-7% of all NSCLC patients and result in the constitutive activation of the ALK signalling pathway, promoting cell proliferation and survival (Figure 1).1,2 From a clinical perspective, ALK-positive NSCLC patients tend to be younger, generally don’t have a smoking history, and most commonly present with an adenocarcinoma histology.1,2 The therapeutic revolution for patients with ALK-positive metastatic NSCLC (mNSCLC) kicked off with the introduction of crizotinib. Prior to this, ALK-rearranged mNSCLC patients were treated with chemotherapy, resulting in a median overall survival (OS) of about 15 months.3 In 2014, results of the phase III PROFILE 1014 study demonstrated the superiority of crizotinib over platinum-pemetrexed in the first line treatment of patients with ALK-positive mNSCLC, both in terms of progression-free survival (PFS; median: 10.9 vs. 7 months; HR[95%CI]: 0.45[0.35-0.60]) and objective response rate (ORR; 74% vs. 45%, p< 0.001).4 These convincing data set the benchmark for future clinical trials and established crizotinib as the new standard of care for newly diagnosed ALK-positive mNSCLC patients.5 However, the durability of the responses to crizotinib is limited due to development of acquired resistance and as a result of the poor blood-brain barrier penetration of crizotinib.6 To overcome these limitations, as of 2016 more potent second generation ALK inhibitors (i.e., alectinib, ceritinib, brigatinib) became available with a better penetration into the central nervous system (CNS). These agents initially proved their worth in patients who progressed on crizotinib but were subsequently also shown to significantly outperform crizotinib in newly diagnosed patients. As a result, these agents have gradually replaced crizotinib as the preferred first line treatment for patients with ALK-positive mNSCLC.7-12
Figure 1. Incidence and variety of oncogenic driver mutations in mNSCLC.20
When using the second-generation ALK inhibitors mentioned above, the median PFS of patients with newly diagnosed ALK-positive mNSCLC markedly improved compared to the crizotinib era, with a median PFS ranging from 17-35 months and a 3-year PFS rate of 43-46%.10-12 However, the development of acquired resistance and intracranial progression remains to be problematic. Lorlatinib is a third generation ALK inhibitor with a distinct molecular structure that facilitates a better crossing of the blood-brain barrier. Furthermore, lorlatinib is more potent against wild-type ALK, with a greater coverage of ALK resistance mutations than second-generation ALK inhibitors.13
Based on the results of a phase I/II study, lorlatinib was initially approved for the treatment of ALK-positive mNSCLC who progressed on a second-generation ALK inhibitor.13 However, this study also included a cohort of newly-diagnosed patients in whom lorlatinib also exhibited very promising activity.13 This formed the basis for the phase III CROWN trial in which lorlatinib was compared head-to-head to crizotinib in the treatment of previously untreated, advanced, ALK-positive NSCLC.14,15
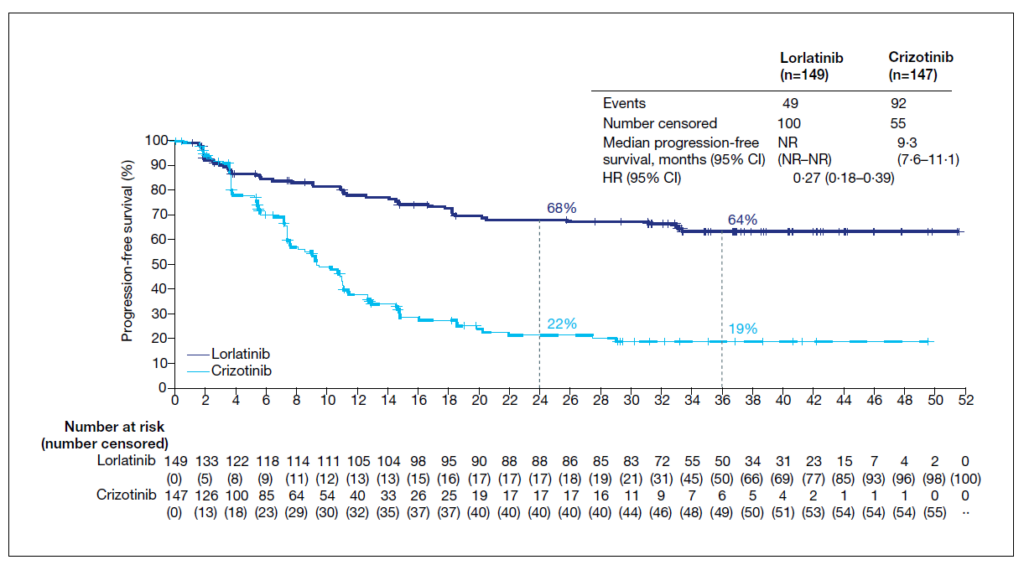
The CROWN trial randomized a total of 296 patients with ALK-positive advanced NSCLC who did not receive prior systemic therapy in the metastatic setting to receive lorlatinib (100 mg daily) or crizotinib (250 mg bid) in 28-day cycles. To be eligible for the study, patients had to have an ECOG performance score of 0-2 and have at least one extracranial measurable target lesion. Importantly, also patients with asymptomatic treated or untreated brain metastases were eligible for inclusion.14 After a median follow-up of 36.7 months, the median PFS for patients in the lorlatinib arm was still not reached, while this was reported at 9.3 months for crizotinib (HR[95%CI]: 0.27[0.18-0.39). At the three-year landmark, this translates into a PFS rate of 64% with lorlatinib, which is more than threefold the 19% PFS rate observed with crizotinib (Figure 2). As such, the PFS obtained with first line lorlatinib exceeds the PFS reported in other trials of ALK TKIs in patients with mNSCLC or in any phase III trial evaluating a targeted therapy in this setting.10-12,15 On top of that, the significant PFS benefit of lorlatinib over crizotinib was maintained across all investigated subgroups, including patients with baseline brain metastases (median PFS not reached vs. 7.2 months; HR[95%CI]: 0.21[0.10-0.44]). Among patients with baseline brain metastases, 68% of patients in the lorlatinib arm were alive and free of progression after three years as compared to 23% in the crizotinib arm. Also in terms of ORR lorlatinib proved to be significantly better than crizotinib, with an ORR of 77% and 59%, respectively (OR[95%CI]: 2.37[1.41-4.10]).15
Figure 2. PFS with lorlatinib vs. crizotinib as first line treatment for ALK-positive mNSCLC.15
The results of the phase III CROWN trial also underscore the excellent CNS activity of lorlatinib. In fact, the proportion of patients without intracranial progression at 3 years was 92% with lorlatinib as compared to 38% with crizotinib. Specifically looking at patients without baseline brain metastases, the probability of being free of intracranial progression at three years was 99% (N= 111/112) with lorlatinib as compared to 50% with crizotinib.15 This clearly suggests a protective effect of lorlatinib against the development of brain metastases. Apart from protecting against the development of (new) brain metastases, lorlatinib also proved to be effective against existing CNS lesions. In fact, the confirmed intracranial ORR with lorlatinib among patients with brain metastases at baseline was reported at 83.3%, with a complete intracranial response in 72% of patients. In contrast, the corresponding rates with crizotinib were 23% and 8%, respectively.15 This excellent CNS activity of lorlatinib is of special clinical relevance given the high incidence of brain involvement in this disease entity. Indeed, data show that 24–42% of all ALK-positive mNSCLC patients are diagnosed with brain metastases at the time of diagnosis, with half of the patients developing brain metastases within the next three 3 years.16
The incidence of grade 3-4 adverse events (AEs) with lorlatinib was reported at 76%, which is higher than the 57% incidence seen with crizotinib. This difference was mainly driven by a higher incidence of grade 3/4 alterations in lipid concentrations. These events, including hypercholesterolaemia and hyperlipidaemia, were typically asymptomatic and readily managed with lipid-lowering agents or drug discontinuations or dose reduction. Importantly, this higher incidence of grade 3/4 AEs did not lead to a higher incidence of treatment discontinuation with lorlatinib compared to crizotinib (7% vs. 10%). In the lorlatinib group, AEs leading to dose reductions were reported in 32 (21%) patients.15 The increased CNS-penetration of lorlatinib also leads to a higher rate of CNS toxicity compared to crizotinib. CNS AEs occurred in 39% of the lorlatinib group, but these events rarely exceeded grade 1/2 in severity.15 Only 2 out of 11 treatment discontinuations in CROWN were due to CNS AEs. Most (62%) CNS AEs were managed without intervention and 23% were managed with lorlatinib dose modification (reduction and/or interruption, with or without concomitant medication).17 Of note, landmark analysis of PFS showed that lorlatinib dose reductions (i.e., to 75mg or 50mg once daily) don’t have an impact on the treatment efficacy.17 The manageable safety profile of lorlatinib is also reflected by the quality of life (QoL) data in the CROWN trial. In fact, a post-hoc analysis of this trial shows that the median time to deterioration in global QoL was 24.7 months with lorlatinib as compared to 12.0 months with crizotinib.15 Furthermore, the occurrence of CNS AEs did not result in a clinically meaningful difference in patient-reported quality of life.17
The convincing results of the phase III CROWN trial add further support for the use of next-generation ALK inhibitors as the preferred first-line treatment for patients with ALK-positive mNSCLC.15 This statement has also been adopted by the European Society of Medical Oncology in their most recent treatment guidelines for patients with oncogene-driven mNSCLC (ESMO-MCBS v1.1 score 4).12,18 A recent Cochrane Review on this matter brings further weight to this by showing a superior PFS and OS with next-generation ALK inhibitors (alectinib, brigatinib and lorlatinib) compared to crizotinib as first-line treatment for patients with ALK-positive mNSCLC.19 Specifically, for lorlatinib, the recently reported long-term efficacy data on the use of this agent in first-line compare favourably to long-term data with second-generation ALK TKIs.10,11,15 In this, however, we need to underscore that cross-trial comparisons have important limitations, and no clear conclusion can be made in the absence of a head-to-head comparison. The CROWN study also demonstrated high intracranial response rates with lorlatinib, with a beneficial effect on time to intracranial progression. This supports the use of lorlatinib in the first line treatment of patients with established brain metastases and in the prevention of CNS involvement in patients without brain metastases.15,17 Importantly, the occurrence of CNS AEs does not result in a clinically meaningful difference in patient reported QoL.17 As such, lorlatinib proves to be a highly effective treatment option, with a manageable safety profile, for treatment-naive patients with advanced, ALK-positive NSCLC, irrespective of the presence of brain metastases.15 As of May 1st, 2023, lorlatinib is reimbursed in Belgium in this setting.
An Expert Opinion – Dr. Christophe Compère (CHIREC Cancer Institue)
The treatment landscape for patients with ALK-positive NSCLC is continuously evolving and to date we have several efficient second-generation ALK-inhibitors that result in long-term PFS. However, third generation ALK-inhibitors are entering the market and challenge us, as lung cancer specialists, with better treatment outcomes, especially for those patients with brain metastases. In the upcoming months, the availability of lorlatinib in first line will probably prove to be practice-changing in our daily clinical practice. The three-year follow up results from the CROWN study are now available and are – at least in my opinion – very exciting. In terms of efficacy, the data speak for themselves; the PFS data are very encouraging and I’m now eagerly awaiting the OS data that will probably be communicated soon. Side effects of lorlatinib are certainly manageable and are in part similar to those of the older generation ALK-inhibitors. However, there are also some differences. The most important side effect of lorlatinib is hyperlipidaemia, which is treatable. Some patients also experience liver problems, but these are usually linked to drug-drug interactions and can thus often be managed by drug substitutions. Furthermore, patients may experience oedema or central nervous system disorders, such as confusion, depression, or hallucination. The answer to these side-effects usually lies in a dose reduction. However, it is interesting to know that in the CROWN trial, approximately 50% of patients had to interrupt treatment for a short time, but this was highly comparable in the lorlatinib and crizotinib arms. Data with lorlatinib also show that this agent effectively prevents the development of CNS metastases and comes with a pronounced intracranial activity in patients with brain metastases at baseline.In my opinion, if overall survival data confirm the current findings, lorlatinib is profiling itself to become the gold-standard in the first-line treatment of patients with ALK-positive mNSCLC.
References
Abbreviations: AE; adverse event, ALK; anaplastic lymphoma kinase, Bid; twice daily, CNS; central nervous system, ECOG; Eastern Cooperative Oncology Group, ESMO; European Society of Medical Oncology, HR; hazard ratio, MCBS; magnitude of clinical benefit scale, NSCLC; non-small cell lung cancer, ORR; objective response rate, OS; overall survival, PFS; progression-free survival, QoL; quality of life, TKI; tyrosine kinase inhibitor.
MAY 2023/230447
▼This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions. NAME OF THE MEDICINAL PRODUCT: Lorviqua 25 mg and 100 mg film‑coated tablets. QUALITATIVE AND QUANTITATIVE COMPOSITION: Lorviqua 25 mg resp. 100 mg film‑coated tablets: Each film‑coated tablet contains 25 mg resp. 100 mg of lorlatinib. Excipient with known effect: Each film‑coated tablet contains 1.58 mg resp. 4.20 mg of lactose monohydrate. PHARMACEUTICAL FORM: Film‑coated tablet (tablet). Lorviqua 25 mg film‑coated tablets: Round (8 mm) light pink immediate release film‑coated tablet, debossed with “Pfizer” on one side and “25” and “LLN” on the other side. Lorviqua 100 mg film‑coated tablets: Oval (8.5 × 17 mm) dark pink immediate release film‑coated tablet, debossed with “Pfizer” on one side and “LLN 100” on the other side. CLINICAL PARTICULARS: Therapeutic indications: Lorviqua as monotherapy is indicated for the treatment of adult patients with anaplastic lymphoma kinase (ALK)‑positive advanced non‑small cell lung cancer (NSCLC) previously not treated with an ALK inhibitor. Lorviqua as monotherapy is indicated for the treatment of adult patients with ALK‑positive advanced NSCLC whose disease has progressed after: alectinib or ceritinib as the first ALK tyrosine kinase inhibitor (TKI) therapy; or crizotinib and at least one other ALK TKI.. Posology and method of administration: Treatment with lorlatinib should be initiated and supervised by a physician experienced in the use of anticancer medicinal products. Detection of ALK‑positive NSCLC is necessary for selection of patients for treatment with lorlatinib because these are the only patients for whom benefit has been shown. Assessment for ALK‑positive NSCLC should be performed by laboratories with demonstrated proficiency in the specific technology being utilised. Improper assay performance can lead to unreliable test results. Posology: The recommended dose is 100 mg lorlatinib taken orally once daily. Duration of treatment: Treatment with lorlatinib should be continued until disease progression or unacceptable toxicity. Delayed or missed doses: If a dose of Lorviqua is missed, then it should be taken as soon as the patient remembers unless it is less than 4 hours before the next dose, in which case the patient should not take the missed dose. Patients should not take 2 doses at the same time to make up for a missed dose. Dose modifications: Dosing interruption or dose reduction may be required based on individual safety and tolerability. Lorlatinib dose reduction levels are summarised below: First dose reduction: 75 mg taken orally once daily; Second dose reduction: 50 mg taken orally once daily. Lorlatinib should be permanently discontinued if the patient is unable to tolerate the 50 mg dose taken orally once daily. Dose modification recommendations for toxicities and for patients who develop atrioventricular (AV) block are provided in Table 1. Table 1. Recommended lorlatinib dose modifications for adverse reactions: Hypercholesterolaemia or hypertriglyceridaemia: Mild hypercholesterolaemia (cholesterol between ULN and 300 mg/dL or between ULN and 7.75 mmol/L) OR Moderate hypercholesterolaemia (cholesterol between 301 and 400 mg/dL or between 7.76 and 10.34 mmol/L) OR Mild hypertriglyceridaemia (triglycerides between 150 and 300 mg/dL or 1.71 and 3.42 mmol/L) OR Moderate hypertriglyceridaemia (triglycerides between 301 and 500 mg/dL or 3.43 and 5.7 mmol/L): Introduce or modify lipid lowering therapyb in accordance with respective prescribing information; continue lorlatinib at same dose. Severe hypercholesterolaemia (cholesterol between 401 and 500 mg/dL or between 10.35 and 12.92 mmol/L) OR Severe hypertriglyceridaemia (triglycerides between 501 and 1,000 mg/dL or 5.71 and 11.4 mmol/L): Introduce the use of lipid lowering therapyb; if currently on lipid lowering therapy, increase the dose of this therapyb in accordance with respective prescribing information; or change to a new lipid lowering therapyb. Continue lorlatinib at the same dose without interruption. Life threatening hypercholesterolaemia (cholesterol over 500 mg/dL or over 12.92 mmol/L) OR Life threatening hypertriglyceridaemia (triglycerides over 1,000 mg/dL or over 11.4 mmol/L): Introduce the use of lipid lowering therapyb or increase the dose of this therapyb in accordance with respective prescribing information or change to a new lipid lowering therapyb. Withhold lorlatinib until recovery of hypercholesterolaemia and/or hypertriglyceridaemia to moderate or mild severity grade. Re-challenge at same lorlatinib dose while maximising lipid lowering therapyb in accordance with respective prescribing information. If severe hypercholesterolaemia and/or hypertriglyceridaemia recur despite maximal lipid lowering therapyb in accordance with respective prescribing information, reduce lorlatinib by 1 dose level. Central nervous system effects (CNS) (comprises psychotic effects and changes in cognition, mood, mental status or speech): Grade 2: Moderate OR Grade 3: Severe: Withhold dose until toxicity is less than or equal to Grade 1. Then resume lorlatinib at 1 reduced dose level. Grade 4: Life threatening/Urgent intervention indicated: Permanently discontinue lorlatinib. Lipase/Amylase increase: Grade 3: Severe OR Grade 4: Life threatening/Urgent intervention indicated: Withhold lorlatinib until lipase or amylase returns to baseline. Then resume lorlatinib at 1 reduced dose level. Interstitial lung disease (ILD)/Pneumonitis: Grade 1: Mild OR Grade 2: Moderate: Withhold lorlatinib until symptoms have returned to baseline and consider initiating corticosteroids. Resume lorlatinib at 1 reduced dose level. Permanently discontinue lorlatinib if ILD/pneumonitis recurs or fails to recover after 6 weeks of lorlatinib hold and steroid treatment. Grade 3: Severe OR Grade 4: Life threatening/Urgent intervention indicated: Permanently discontinue lorlatinib. PR interval prolongation/Atrioventricular (AV) block: First degree AV block: Asymptomatic: Continue lorlatinib at the same dose without interruption. Consider effects of concomitant medicinal products, and assess and correct electrolyte imbalance that may prolong PR interval. Monitor ECG/symptoms potentially related to AV block closely. First degree AV block: Symptomatic: Withhold lorlatinib. Consider effects of concomitant medicinal products, and assess and correct electrolyte imbalance that may prolong PR interval. Monitor ECG/symptoms potentially related to AV block closely. If symptoms resolve, resume lorlatinib at 1 reduced dose level. Second degree AV block Asymptomatic: Withhold lorlatinib. Consider effects of concomitant medicinal products, and assess and correct electrolyte imbalance that may prolong PR interval. Monitor ECG/symptoms potentially related to AV block closely. If subsequent ECG does not show second degree AV block, resume lorlatinib at 1 reduced dose level. Second degree AV block Symptomatic: Withhold lorlatinib. Consider effects of concomitant medicinal products, and assess and correct electrolyte imbalance that may prolong PR interval. Refer for cardiac observation and monitoring. Consider pacemaker placement if symptomatic AV block persists. If symptoms and the second degree AV block resolve or if patients revert to asymptomatic first degree AV block, resume lorlatinib at 1 reduced dose level. Complete AV block: Withhold lorlatinib. Consider effects of concomitant medicinal products, and assess and correct electrolyte imbalance that may prolong PR interval. Refer for cardiac observation and monitoring. Pacemaker placement may be indicated for severe symptoms associated with AV block. If AV block does not resolve, placement of a permanent pacemaker may be considered. If pacemaker placed, resume lorlatinib at full dose. If no pacemaker placed, resume lorlatinib at 1 reduced dose level only when symptoms resolve and PR interval is less than 200 msec. Hypertension: Grade 3 (SBP greater than or equal to 160 mmHg or DBP greater than or equal to 100 mmHg; medical intervention indicated; more than one antihypertensive drug, or more intensive therapy than previously used indicated): Withhold lorlatinib until hypertension has recovered to Grade 1 or less (SBP less than 140 mmHg and DBP less than 90 mmHg), then resume lorlatinib at the same dose. If Grade 3 hypertension recurs, withhold lorlatinib until recovery to Grade 1 or less, and resume at a reduced dose. If adequate hypertension control cannot be achieved with optimal medical management, permanently discontinue lorlatinib. Grade 4 (Life-threatening consequences, urgent intervention indicated): Withhold lorlatinib until recovery to Grade 1 or less, and resume at a reduced dose or permanently discontinue lorlatinib. If Grade 4 hypertension recurs, permanently discontinue lorlatinib. Hyperglycaemia: Grade 3 OR Grade 4 (Persistent hyperglycaemia greater than 250 mg/dL despite optimal anti hyperglycaemic therapy): Withhold lorlatinib until hyperglycaemia is adequately controlled, then resume lorlatinib at the next lower dosage. If adequate hyperglycaemic control cannot be achieved with optimal medical management, permanently discontinue lorlatinib. Other adverse reactions: Grade 1: Mild OR Grade 2: Moderate: Consider no dose modification or reduce by 1 dose level, as clinically indicated. Greater than or equal to Grade 3: Severe: Withhold lorlatinib until symptoms resolve to less than or equal to Grade 2 or baseline. Then resume lorlatinib at 1 reduced dose level. Abbreviations: CNS=central nervous system; CTCAE=Common Terminology Criteria for Adverse Events; DBP=diastolic blood pressure; ECG=electrocardiogram; HMG CoA=3 hydroxy 3 methylglutaryl coenzyme A; NCI=National Cancer Institute; SBP=systolic blood pressure; ULN=upper limit of normal. aGrade categories are based on NCI CTCAE classifications. bLipid lowering therapy may include: HMG CoA reductase inhibitor, nicotinic acid, fibric acid derivatives, or ethyl esters of omega 3 fatty acids. Strong cytochrome P‑450 (CYP) 3A4/5 inhibitors: Concurrent use of lorlatinib with medicinal products that are strong CYP3A4/5 inhibitors and grapefruit juice products may increase lorlatinib plasma concentrations. An alternative concomitant medicinal product with less potential to inhibit CYP3A4/5 should be considered. If a strong CYP3A4/5 inhibitor must be co‑administered, the starting lorlatinib dose of 100 mg once daily should be reduced to once daily 75 mg dose. If concurrent use of the strong CYP3A4/5 inhibitor is discontinued, lorlatinib should be resumed at the dose used prior to the initiation of the strong CYP3A4/5 inhibitor and after a washout period of 3 to 5 half‑lives of the strong CYP3A4/5 inhibitor. Special populations: Elderly (≥ 65 years): Due to the limited data on this population, no dose recommendation can be made for patients aged 65 years and older. Renal impairment: No dose adjustment is needed for patients with normal renal function and mild or moderate renal impairment [absolute estimated glomerular filtration rate (eGFR): ≥ 30 mL/min]. A reduced dose of lorlatinib is recommended in patients with severe renal impairment (absolute eGFR < 30 mL/min), e.g. a once daily starting dose of 75 mg taken orally. No information is available for patients on renal dialysis. Hepatic impairment: No dose adjustments are recommended for patients with mild hepatic impairment. No information is available for lorlatinib in patients with moderate or severe hepatic impairment. Therefore, lorlatinib is not recommended in patients with moderate to severe hepatic impairment. Paediatric population: The safety and efficacy of lorlatinib in paediatric patients below 18 years have not been established. No data are available. Method of administration: Lorviqua is for oral use. Patients should be encouraged to take their dose of lorlatinib at approximately the same time each day with or without food. The tablets should be swallowed whole (tablets should not be chewed, crushed or split prior to swallowing). No tablet should be ingested if it is broken, cracked, or otherwise not intact. Contraindications: Hypersensitivity to lorlatinib or to any of the excipients listed. Concomitant use of strong CYP3A4/5 inducers.
Special warnings and precautions for use: Hyperlipidaemia: The use of lorlatinib has been associated with increases in serum cholesterol and triglycerides. Median time of occurrence of severe increase in serum cholesterol and triglycerides is 104 days (range: 29 to 518 days) and 120 days (range: 15 to 780 days), respectively. Serum cholesterol and triglycerides should be monitored before initiation of lorlatinib; 2, 4 and 8 weeks after initiating lorlatinib; and regularly thereafter. Initiate or increase the dose of lipid‑lowering medicinal products, if indicated. Central nervous system effects: Central nervous system (CNS) effects have been observed in patients receiving lorlatinib, including psychotic effects and changes in cognitive function, mood, mental status or speech . Dose modification or discontinuation may be required for those patients who develop CNS effects. Atrioventricular block: Lorlatinib was studied in a population of patients that excluded those with second‑degree or third‑degree AV block (unless paced) or any AV block with PR interval > 220 msec. PR interval prolongation and AV block have been reported in patients receiving lorlatinib. Monitor electrocardiogram (ECG) prior to initiating lorlatinib and monthly thereafter, particularly in patients with predisposing conditions to the occurrence of clinically significant cardiac events. Dose modification may be required for those patients who develop AV block. Left ventricular ejection fraction decrease: Left ventricular ejection fraction (LVEF) decrease has been reported in patients receiving lorlatinib who had baseline and at least one follow-up LVEF assessment. Based on the available clinical study data, it is not possible to determine a causal relationship between effects on changes in cardiac contractility and lorlatinib. In patients with cardiac risk factors and those with conditions that can affect LVEF, cardiac monitoring, including LVEF assessment at baseline and during treatment, should be considered. In patients who develop relevant cardiac signs/symptoms during treatment, cardiac monitoring, including LVEF assessment, should be considered. Lipase and amylase increase: Elevations of lipase and/or amylase have occurred in patients receiving lorlatinib. Median time of occurrence of increase in serum lipase and amylase is 141 days (range: 1 to 1091 days) and 138 days (range: 1 to 1112 days), respectively. Risk of pancreatitis should be considered in patients receiving lorlatinib due to concomitant hypertriglyceridemia and/or a potential intrinsic mechanism. Patients should be monitored for lipase and amylase elevations prior to the start of lorlatinib treatment and regularly thereafter as clinically indicated. Interstitial lung disease/Pneumonitis: Severe or life‑threatening pulmonary adverse reactions consistent with ILD/pneumonitis have occurred with lorlatinib . Any patient who presents with worsening of respiratory symptoms indicative of ILD/pneumonitis (e.g. dyspnoea, cough and fever) should be promptly evaluated for ILD/pneumonitis. Lorlatinib should be withheld and/or permanently discontinued based on severity. Hypertension: Hypertension has been reported in patients receiving lorlatinib . Blood pressure should be controlled prior to initiation of lorlatinib. Blood pressure should be monitored after 2 weeks and at least monthly thereafter during treatment with lorlatinib. Lorlatinib should be withheld and resumed at a reduced dose or permanently discontinued based on severity. Hyperglycaemia: Hyperglycaemia has occurred in patients receiving lorlatinib . Fasting serum glucose should be assessed prior to initiation of lorlatinib and monitored periodically thereafter according to national guidelines. Lorlatinib should be withheld and resumed at a reduced dose or permanently discontinued based on severity. Drug‑drug interactions: In a study conducted in healthy volunteers, the concomitant use of lorlatinib and rifampin, a strong CYP3A4/5 inducer, was associated with increases of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) with no increase of total bilirubin and alkaline phosphatase. Concomitant use of a strong CYP3A4/5 inducer is contraindicated. No clinically meaningful changes in liver function tests were seen in healthy subjects after receiving a combination of lorlatinib with the moderate CYP3A4/5 inducer modafinil. Concurrent administration of lorlatinib with CYP3A4/5 substrates with narrow therapeutic indices, including but not limited to alfentanil, ciclosporin, dihydroergotamine, ergotamine, fentanyl, hormonal contraceptives, pimozide, quinidine, sirolimus and tacrolimus, should be avoided since the concentration of these medicinal products may be reduced by lorlatinib. Fertility and pregnancy: During treatment with lorlatinib and for at least 14 weeks after the final dose, male patients with female partners of childbearing potential must use effective contraception, including a condom, and male patients with pregnant partners must use condoms. Male fertility may be compromised during treatment with lorlatinib. Men should seek advice on effective fertility preservation before treatment. Women of childbearing potential should be advised to avoid becoming pregnant while receiving lorlatinib. A highly effective non‑hormonal method of contraception is required for female patients during treatment with lorlatinib, because lorlatinib can render hormonal contraceptives ineffective. If a hormonal method of contraception is unavoidable, then a condom must be used in combination with the hormonal method. Effective contraception must be continued for at least 35 days after completing therapy . It is not known whether lorlatinib affects female fertility. Lactose intolerance: This medicinal product contains lactose as an excipient. Patients with rare hereditary problems of galactose intolerance, total lactase deficiency, or glucose‑galactose malabsorption should not take this medicinal product. Dietary sodium: This medicinal product contains less than 1 mmol sodium (23 mg) per 25 mg or 100 mg tablet. Patients on low sodium diets should be informed that this product is essentially “sodium‑free”.
Interaction with other medicinal products and other forms of interaction: Pharmacokinetic interactions: In vitro data indicate that lorlatinib is primarily metabolised by CYP3A4 and uridine diphosphate‑glucuronosyltransferase (UGT)1A4, with minor contributions from CYP2C8, CYP2C19, CYP3A5 and UGT1A3. Effect of medicinal products on lorlatinib: CYP3A4/5 inducers: Rifampin, a strong inducer of CYP3A4/5, administered at oral doses of 600 mg once daily for 12 days, reduced the mean lorlatinib area under curve (AUCinf) by 85% and Cmax by 76% of a single 100 mg oral dose of lorlatinib in healthy volunteers; increases in AST and ALT were also observed. Concomitant administration of lorlatinib with strong CYP3A4/5 inducers (e.g. rifampicin, carbamazepine, enzalutamide, mitotane, phenytoin and St. John’s wort) may decrease lorlatinib plasma concentrations. The use of a strong CYP3A4/5 inducer with lorlatinib is contraindicated. No clinically meaningful changes in liver function test results were seen after administration of the combination of a single 100 mg oral dose of lorlatinib with the moderate CYP3A4/5 inducer, modafinil (400 mg once daily for 19 days) in healthy volunteers. Concomitant use of modafinil did not have a clinically meaningful effect on lorlatinib pharmacokinetics. CYP3A4/5 inhibitors: Itraconazole, a strong inhibitor of CYP3A4/5, administered at oral doses of 200 mg once daily for 5 days, increased the mean lorlatinib AUCinf by 42% and Cmax by 24% of a single 100 mg oral dose of lorlatinib in healthy volunteers. Concomitant administration of lorlatinib with strong CYP3A4/5 inhibitors (e.g. boceprevir, cobicistat, itraconazole, ketoconazole, posaconazole, troleandomycin, voriconazole, ritonavir, paritaprevir in combination with ritonavir and ombitasvir and/or dasabuvir, and ritonavir in combination with either elvitegravir, indinavir, lopinavir or tipranavir) may increase lorlatinib plasma concentrations. Grapefruit products may also increase lorlatinib plasma concentrations and should be avoided. An alternative concomitant medicinal product with less potential to inhibit CYP3A4/5 should be considered. If a strong CYP3A4/5 inhibitor must be concomitantly administered, a dose reduction of lorlatinib is recommended.Effect of lorlatinib on other medicinal products: CYP3A4/5 substrates: In vitro studies indicated that lorlatinib is a time‑dependent inhibitor as well as an inducer of CYP3A4/5. Lorlatinib 150 mg orally once daily for 15 days decreased AUCinf and Cmax of a single oral 2 mg dose of midazolam (a sensitive CYP3A substrate) by 61% by 50%, respectively; hence, lorlatinib is a moderate CYP3A inducer. Thus, concurrent administration of lorlatinib with CYP3A4/5 substrates with narrow therapeutic indices, including but not limited to alfentanil, ciclosporin, dihydroergotamine, ergotamine, fentanyl, hormonal contraceptives, pimozide, quinidine, sirolimus and tacrolimus, should be avoided since the concentration of these medicinal products may be reduced by lorlatinib. CYP2B6 substrates: Lorlatinib 100 mg once daily for 15 days decreased AUCinf and Cmax of a single oral 100 mg dose of bupropion (a combined CYP2B6 and CYP3A4 substrate) by 49.5% and 53%, respectively. Thus, lorlatinib is a weak inducer of CYP2B6, and no dose adjustment is necessary when lorlatinib is used in combination with medicinal products that are mainly metabolised by CYP2B6. CYP2C9 substrates: Lorlatinib 100 mg once daily for 15 days decreased AUCinf and Cmax of a single oral 500 mg dose of tolbutamide (a sensitive CYP2C9 substrate) by 43% and 15%, respectively. Thus, lorlatinib is a weak inducer of CYP2C9, and no dose adjustment is required for medicinal products that are mainly metabolised by CYP2C9. However, patients should be monitored in case of concomitant treatment with medicinal products with narrow therapeutic indices metabolised by CYP2C9 (e.g. coumarin anticoagulants). UGT substrates: Lorlatinib 100 mg once daily for 15 days decreased AUCinf and Cmax of a single oral 500 mg dose of acetaminophen (a UGT, SULT and CYP1A2, 2A6, 2D6, and 3A4 substrate) by 45% and 28%, respectively. Thus, lorlatinib is a weak inducer of UGT, and no dose adjustment is required for medicinal products that are mainly metabolised by UGT. However, patients should be monitored in case of concomitant treatment with medicinal products with narrow therapeutic indices metabolised by UGT. P‑glycoprotein substrates: Lorlatinib 100 mg once daily for 15 days decreased AUCinf and Cmax of a single oral dose of 60 mg fexofenadine [a sensitive P‑glycoprotein (P‑gp) substrate] by 67% and 63%, respectively. Thus, lorlatinib is a moderate inducer of P‑gp. Medicinal products that are P‑gp substrates with narrow therapeutic indices (e.g. digoxin, dabigatran etexilate) should be used with caution in combination with lorlatinib due to the likelihood of reduced plasma concentrations of these substrates. In vitro inhibition and induction studies of other CYP enzymes: In vitro, lorlatinib has a low potential to cause drug‑drug interactions by induction of CYP1A2. In vitro studies with drug transporters other than P‑gp: In vitro studies indicated that lorlatinib may have the potential to inhibit BCRP (gastrointestinal tract), OATP1B1, OATP1B3, OCT1, MATE1 and OAT3 at clinically relevant concentrations. Lorlatinib should be used with caution in combination with substrates of BCRP, OATP1B1, OATP1B3, OCT1, MATE1 and OAT3 as clinically relevant changes in the plasma exposure of these substrates cannot be ruled out.
Undesirable effects: Summary of the safety profile: The most frequently reported adverse reactions were hypercholesterolaemia (81.1%), hypertriglyceridaemia (67.2%), oedema (55.7%), peripheral neuropathy (43.7%), weight increased (30.9%), cognitive effects (27.7%), fatigue (27.3%), arthralgia (23.5%), diarrhoea (22.9%) and mood effects (21.0%). Serious adverse reactions were reported in 7.4% of patients receiving lorlatinib. The most frequent serious adverse drug reactions were cognitive effects and pneumonitis. Dose reductions due to adverse reactions occurred in 20.0% of patients receiving lorlatinib. The most common adverse reactions that led to dose reductions were oedema and peripheral neuropathy. Permanent treatment discontinuation associated with adverse reactions occurred in 3.2% of patients receiving lorlatinib. The most frequent adverse reactions that led to permanent discontinuations were cognitive effects peripheral neuropathy, pneumonitis and psychotic effects. Tabulated list of adverse reactions: Table 2 presents adverse reactions occurring in 476 adult patients treated with lorlatinib 100 mg once daily with advanced NSCLC from Study A (N=327) and CROWN study (N=149). The adverse reactions listed in Table 2 are presented by system organ class and frequency categories, defined using the following convention: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), very rare (< 1/10,000). Within each frequency grouping, undesirable effects are presented in order of decreasing medical seriousness, mentioning: System organ class and adverse reaction: Frequency category, All Grades %, Grades 3-4 %. Table 2. Adverse reactions: Blood and lymphatic system disorders: Anaemia: Very common, 18.5, 4.2. Metabolism and nutrition disorders: Hypercholesterolaemiaa: Very common, 81.1, 18.3; Hypertriglyceridaemiab: Very common, 67.2, 19.3. Hyperglycaemia: Common: 9.2, 3.2. Psychiatric disorders: Mood effectsc: Very common, 21.0, 1.5; Psychotic effectsd: Common, 6.5, 0.4; Mental status changes: Common, 2.0, 1.7. Nervous system disorders: Cognitive effectse: Very common, 27.7, 2.9; Peripheral neuropathyf: Very common, 43.7, 2.7; Headache: Very common, 17.9, 0.6; Speech effectsg: Common, 8.2, 0.6. Eye disorders: Vision disorderh: Very common, 17.2, 0.2. Vascular disorders: Hypertension: Very common: 13.0, 6.1. Respiratory, thoracic and mediastinal disorders: Pneumonitisi: Common, 1.9, 0.6. Gastrointestinal disorders: Diarrhoea: Very common, 22.9, 1.5; Nausea: Very common, 17.6, 0.6; Constipation: Very common, 17.4, 0.2. Skin and subcutaneous tissue disorders: Rashj: Very common, 13.7, 0.2. Musculoskeletal and connective tissue disorders: Arthralgia: Very common, 23.5, 0.8; Myalgiak: Very common, 19.3, 0.2. General disorders and administration site conditions: Oedemal: Very common, 55.7, 2.7; Fatiguem: Very common, 27.3, 1.3. Investigations: Weight increased: Very common, 30.9, 10.1; Lipase increased: Very common, 12.4, 6.9; Amylase increased: Very common, 11.3, 2.7; Electrocardiogram PR prolongation: Uncommon, 0.8, 0. Adverse reactions that represent the same medical concept or condition were grouped together and reported as a single adverse reaction in the table above. Terms actually reported in the studies and contributing to the relevant adverse reaction are indicated in parentheses, as listed below. aHypercholesterolaemia (including blood cholesterol increased, hypercholesterolaemia). bHypertriglyceridaemia (including blood triglycerides increased, hypertriglyceridaemia). cMood effects (including affective disorder, affect lability, aggression, agitation, anger, anxiety, bipolar I disorder, depressed mood, depression, depressive symptom, euphoric mood, irritability, mania, mood altered, mood swings, panic attack, personality change, stress). dPsychotic effects (including auditory hallucination, hallucination, visual hallucination). eCognitive effects (including events from SOC Nervous system disorders: amnesia, cognitive disorder, dementia, disturbance in attention, memory impairment, mental impairment; and also including events from SOC Psychiatric disorders: attention deficit/hyperactivity disorder, confusional state, delirium, disorientation, reading disorder). Within these effects, terms from SOC Nervous system disorders were more frequently reported than terms from SOC Psychiatric disorder. fPeripheral neuropathy (including burning sensation, dysaesthesia, formication, gait disturbance, hypoaesthesia, motor dysfunction, muscular weakness, neuralgia, neuropathy peripheral, neurotoxicity, paraesthesia, peripheral motor neuropathy, peripheral sensory neuropathy, peroneal nerve palsy, sensory disturbance). gSpeech effects (dysarthria, slow speech, speech disorder). hVision disorder (including diplopia, photophobia, photopsia, vision blurred, visual acuity reduced, visual impairment, vitreous floaters). iPneumonitis (including interstitial lung disease, lung opacity, pneumonitis). jRash (including dermatitis acneiform, maculopapular rash, pruritic rash, rash). kMyalgia (including musculoskeletal pain, myalgia). lOedema (including generalised oedema, oedema, oedema peripheral, peripheral swelling, swelling). mFatigue (including asthenia, fatigue). Description of selected adverse reactions: Hypercholesterolaemia/hypertriglyceridaemia: Adverse reactions of increase in serum cholesterol or triglycerides were reported in 81.1% and 67.2% of patients, respectively. Of those, mild or moderate adverse reactions of hypercholesterolaemia or hypertriglyceridaemia occurred in 62.8% and 47.9% of patients, respectively. The median time to onset for both hypercholesterolaemia and hypertriglyceridaemia was 15 days (hypercholesterolaemia range: 1 to 784 days; hypertriglyceridaemia range: 1 to 796 days). The median duration of hypercholesterolaemia and hypertriglyceridaemia was 451 and 427 days, respectively. Central nervous system effects: CNS adverse reactions were primarily cognitive effects (27.7%), mood effects (21.0%), speech effects (8.2%) and psychotic effects (6.5%), and were generally mild, transient, and reversible spontaneously upon dose delay and/or dose reduction . The most frequent cognitive effect of any grade was memory impairment (11.3%), and the most frequent Grade 3 or 4 reactions were confusional state and cognitive disorder (1.7% and 0.8%, respectively). The most frequent mood effect of any grade was anxiety (6.5%), and the most frequent Grade 3 and 4 reactions were irritability and depression (0.8% and 0.4%, respectively). The most frequent speech effect of any grade was dysarthria (4.0%), and the Grade 3 or 4 reactions were dysarthria, slow speech and speech disorder (0.2% each). The most frequent psychotic effect of any grade was hallucination (3.7%) and the most frequent Grade 3 or 4 reactions were hallucination, hallucination auditory and hallucination visual (0.3% each). Median time to onset for cognitive, mood, speech and psychotic effects was 109, 43, 49 and 23 days, respectively. Median duration of cognitive, mood, speech and psychotic effects was 223, 143, 147 and 74 days, respectively. Hypertension: Adverse reactions of hypertension were reported in 13% of patients from Study A and CROWN (B7461006). Of those, mild or moderate adverse reactions of hypertension occurred in 6.9% of patients . The median time to onset of hypertension was 208 days (range: 1 to 1028 days). The median duration of hypertension was 219 days. Hyperglycaemia: Adverse reactions of hyperglycaemia were reported in 9.2% of patients from Study A and CROWN (B7461006). Of those, mild or moderate adverse reactions of hyperglycaemia occurred in 6.1% of patients . The median time to onset of hyperglycaemia was 145 days (range: 1 to 1058 days). The median duration of hyperglycaemia was 113 days. Reporting of suspected adverse reactions: Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the Federal Agency for Drugs and Health Products–Vigilance Department, Galileelaan-Avenue Galilée 5/03, 1210 Brussels (website: www.eenbijwerkingmelden.be-www.notifieruneffetindesirable.be; e-mail: adr@fagg-afmps.be). MARKETING AUTHORISATION HOLDER: Pfizer Europe MA EEIG, Boulevard de la Plaine 17, 1050 Bruxelles, Belgium. MARKETING AUTHORISATION NUMBER(S): EU/1/19/1355/001, EU/1/19/1355/002, EU/1/19/1355/003. Delivery: On medical prescription. DATE OF REVISION OF THE TEXT: 04/2023. Detailed information on this medicinal product is available on the website of the European Medicines Agency http://www.ema.europa.eu.