As of December 1st 2023, KEYTRUDA® (pembrolizumab) is reimbursed as monotherapy for the adjuvant treatment of adults with NSCLC who are at high risk of recurrence following complete resection and platinum-based chemotherapy, regardless of PD-L1 expression. This is based on the positive results of the PEARLS/KEYNOTE-091 trial, which included stage IB [T2a ≥ 4 cm], II or IIIA patients by AJCC 7th edition across all PD-L1 expression levels.1-4
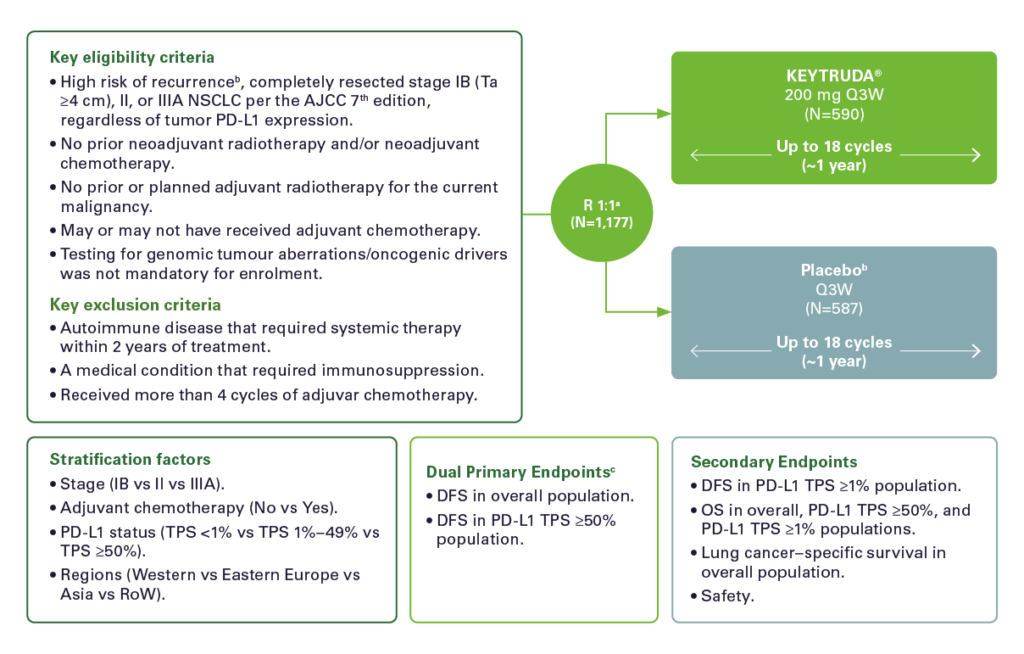
Study Design: 1-3
PEARLS/KEYNOTE-091 is a multicenter, randomized, triple-blind, placebo-controlled trial in patients with resected stage IB (T2a ≥4 cm), II or IIIAa NSCLC, regardless of PD-L1 expression.
Efficacy: 1
At the final analysis for disease-free survival (DFS) and after a median follow-up of 46.7 months, the study demonstrated clinically meaningful DFS with a 24% risk reduction vs. placebo (HR = 0.76 [95% CI: 0.64, 0.91]) in patients who received adjuvant chemotherapy.1
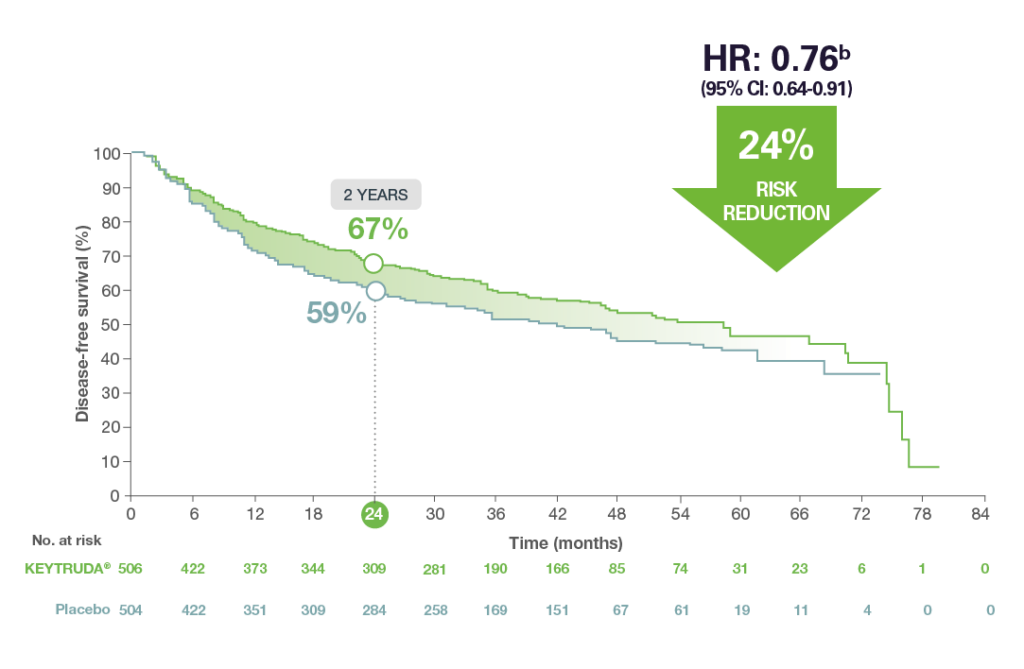
Adapted from the KEYTRUDA® SmPC.1

Safety:1-3
Grade 3 to 5 adverse events of any cause occurred in 34% of patients in the KEYTRUDA® group vs. 26% in the placebo group.1
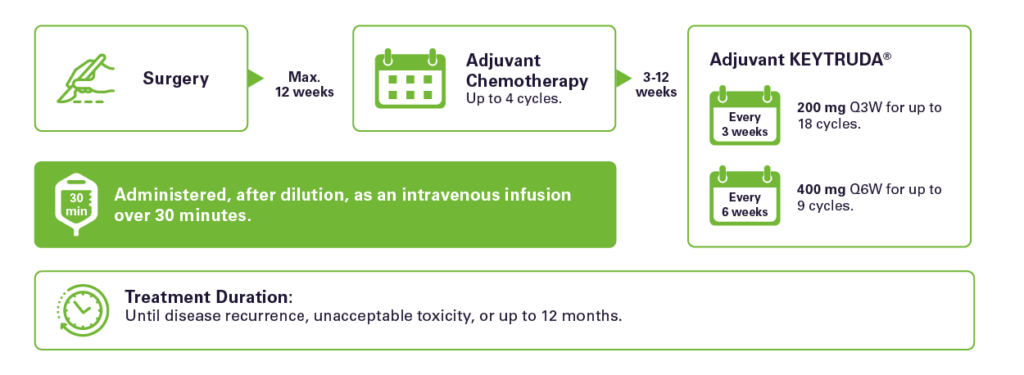
Dosing scheme: 1
More information:
Detailed reimbursement conditions can be consulted on the RIZIV/INAMI website.4
Don’t hesitate to contact your MSD Account Manager if you want to discuss this new indication in more detail or click on the link below to read more about the trial & the results.
Please consult the full prescribing information before prescribing or delivering the product.
Abbreviations:
AJCC = American Joint Committee on Cancer; ASCO = American Society for Clinical Oncology; CI = Confidence Interval; DFS = Disease-Free Survival; HR = Hazard Ratio; INAMI = Institut National d’Assurance Maladie-Invalidité; N = No. = Number; NSCLC = Non-Small Cell Lung Cancer; Oncol = Oncology; OS = Overall Survival; PD-L1 = Programmed Death-Ligand 1; Q3W = 3-weekly scheme; Q6W = 6-weekly scheme; R = Randomized; RIZIV = Rijksinstituut voor Ziekte- en Invaliditeitsverzekering; RoW = Rest of World; T2a = 3-4cm; TPS = Tumor Proportion Score.
References:
Footnotes:
aAs defined by the American Joint Committee on Cancer (AJCC) 7th edition. bThe selection criteria that define patients with high risk of recurrence who are included in the therapeutic indication are reflective of the patient population with stage IB [T2a ≥4 cm], II or IIIA according to the 7th edition staging system; and can be found in section 5.1 of the SmPC.
MSD Belgium BV/SRL, Vorstlaan 25 Boulevard du Souverain, 1170 Brussels
BE-LAM-00324. Date of last revision: 12/2023.
1. NAME OF THE MEDICINAL PRODUCT KEYTRUDA 25 mg/ml concentrate for solution for infusion. 2. QUALITATIVE AND QUANTITATIVE COMPOSITION One vial of 4 ml of concentrate contains 100 mg of pembrolizumab. Each ml of concentrate contains 25 mg of pembrolizumab. Pembrolizumab is a humanised monoclonal anti-programmed cell death‑1 (PD‑1) antibody (IgG4/kappa isotype with a stabilising sequence alteration in the Fc region) produced in Chinese hamster ovary cells by recombinant DNA technology. For the full list of excipients, see section 6.1. 3. PHARMACEUTICAL FORM Concentrate for solution for infusion. Clear to slightly opalescent, colourless to slightly yellow solution, pH 5.2 – 5.8. 4. CLINICAL PARTICULARS 4.1 Therapeutic indications Melanoma KEYTRUDA as monotherapy is indicated for the treatment of adults and adolescents aged 12 years and older with advanced (unresectable or metastatic) melanoma. KEYTRUDA as monotherapy is indicated for the adjuvant treatment of adults and adolescents aged 12 years and older with Stage IIB, IIC or III melanoma and who have undergone complete resection (see section 5.1). Non‑small cell lung carcinoma (NSCLC) KEYTRUDA as monotherapy is indicated for the adjuvant treatment of adults with non-small cell lung carcinoma who are at high risk of recurrence following complete resection and platinum-based chemotherapy (for selection criteria, see section 5.1).KEYTRUDA as monotherapy is indicated for the first-line treatment of metastatic non-small cell lung carcinoma in adults whose tumours express PD-L1 with a ≥50% tumour proportion score (TPS) with no EGFR or ALK positive tumour mutations. KEYTRUDA, in combination with pemetrexed and platinum chemotherapy, is indicated for the first-line treatment of metastatic non-squamous non‑small cell lung carcinoma in adults whose tumours have no EGFR or ALK positive mutations. KEYTRUDA, in combination with carboplatin and either paclitaxel or nab-paclitaxel, is indicated for the first-line treatment of metastatic squamous non‑small cell lung carcinoma in adults. KEYTRUDA as monotherapy is indicated for the treatment of locally advanced or metastatic non‑small cell lung carcinoma in adults whose tumours express PD-L1 with a ≥1% TPS and who have received at least one prior chemotherapy regimen. Patients with EGFR or ALK positive tumour mutations should also have received targeted therapy before receiving KEYTRUDA. Classical Hodgkin lymphoma (cHL) KEYTRUDA as monotherapy is indicated for the treatment of adult and paediatric patients aged 3 years and older with relapsed or refractory classical Hodgkin lymphoma who have failed autologous stem cell transplant (ASCT) or following at least two prior therapies when ASCT is not a treatment option. Urothelial carcinoma KEYTRUDA as monotherapy is indicated for the treatment of locally advanced or metastatic urothelial carcinoma in adults who have received prior platinum-containing chemotherapy (see section 5.1). KEYTRUDA as monotherapy is indicated for the treatment of locally advanced or metastatic urothelial carcinoma in adults who are not eligible for cisplatin-containing chemotherapy and whose tumours express PD-L1 with a combined positive score (CPS) ≥ 10 (see section 5.1). Head and neck squamous cell carcinoma (HNSCC) KEYTRUDA, as monotherapy or in combination with platinum and 5-fluorouracil (5-FU) chemotherapy, is indicated for the first-line treatment of metastatic or unresectable recurrent head and neck squamous cell carcinoma in adults whose tumours express PD-L1 with a CPS ≥ 1 (see section 5.1). KEYTRUDA as monotherapy is indicated for the treatment of recurrent or metastatic head and neck squamous cell carcinoma in adults whose tumours express PD-L1 with a ≥ 50% TPS and progressing on or after platinum-containing chemotherapy (see section 5.1). Renal cell carcinoma (RCC) KEYTRUDA, in combination with axitinib, is indicated for the first-line treatment of advanced renal cell carcinoma in adults (see section 5.1). KEYTRUDA, in combination with lenvatinib, is indicated for the first line treatment of advanced renal cell carcinoma in adults (see section 5.1). KEYTRUDA as monotherapy is indicated for the adjuvant treatment of adults with renal cell carcinoma at increased risk of recurrence following nephrectomy, or following nephrectomy and resection of metastatic lesions (for selection criteria, see section 5.1). Microsatellite instability high (MSI-H) or mismatch repair deficient (dMMR) cancers: Colorectal cancer (CRC) KEYTRUDA as monotherapy is indicated for adults with MSI-H or dMMR colorectal cancer in the following settings: first‑line treatment of metastatic colorectal cancer; treatment of unresectable or metastatic colorectal cancer after previous fluoropyrimidine‑based combination therapy. Non-colorectal cancers KEYTRUDA as monotherapy is indicated for the treatment of the following MSI‑H or dMMR tumours in adults with: advanced or recurrent endometrial carcinoma, who have disease progression on or following prior treatment with a platinum‑containing therapy in any setting and who are not candidates for curative surgery or radiation;unresectable or metastatic gastric, small intestine, or biliary cancer, who have disease progression on or following at least one prior therapy. Oesophageal carcinoma KEYTRUDA, in combination with platinum and fluoropyrimidine based chemotherapy, is indicated for the first-line treatment of locally advanced unresectable or metastatic carcinoma of the oesophagus in adults whose tumours express PD‑L1 with a CPS ≥ 10 (see section 5.1). Triple‑negative breast cancer (TNBC) KEYTRUDA, in combination with chemotherapy as neoadjuvant treatment, and then continued as monotherapy as adjuvant treatment after surgery, is indicated for the treatment of adults with locally advanced, or early‑stage triple‑negative breast cancer at high risk of recurrence (see section 5.1). KEYTRUDA, in combination with chemotherapy, is indicated for the treatment of locally recurrent unresectable or metastatic triple‑negative breast cancer in adults whose tumours express PD‑L1 with a CPS ≥ 10 and who have not received prior chemotherapy for metastatic disease (see section 5.1). Endometrial carcinoma (EC) KEYTRUDA, in combination with lenvatinib, is indicated for the treatment of advanced or recurrent endometrial carcinoma in adults who have disease progression on or following prior treatment with a platinum containing therapy in any setting and who are not candidates for curative surgery or radiation. Cervical cancer KEYTRUDA, in combination with chemotherapy with or without bevacizumab, is indicated for the treatment of persistent, recurrent, or metastatic cervical cancer in adults whose tumours express PD‑L1 with a CPS ≥ 1. Gastric or gastro-oesophageal junction (GEJ) adenocarcinoma KEYTRUDA, in combination with trastuzumab, fluoropyrimidine and platinum-containing chemotherapy, is indicated for the first-line treatment of locally advanced unresectable or metastatic HER2-positive gastric or gastro-oesophageal junction adenocarcinoma in adults whose tumours express PD-L1 with a CPS ≥ 1. KEYTRUDA, in combination with fluoropyrimidine and platinum-containing chemotherapy, is indicated for the first-line treatment of locally advanced unresectable or metastatic HER2-negative gastric or gastro-oesophageal junction adenocarcinoma in adults whose tumours express PD‑L1 with a CPS ≥ 1. Biliary tract carcinoma (BTC). KEYTRUDA, in combination with gemcitabine and cisplatin, is indicated for the first-line treatment of locally advanced unresectable or metastatic biliary tract carcinoma in adults. 4.2 Posology and method of administration Therapy must be initiated and supervised by specialist physicians experienced in the treatment of cancer. PD-L1 testing If specified in the indication, patient selection for treatment with KEYTRUDA based on the tumour expression of PD-L1 should be confirmed by a validated test (see sections 4.1, 4.4, 4.8 and 5.1). MSI/MMR testing If specified in the indication, patient selection for treatment with KEYTRUDA based on MSI‑H/dMMR tumour status should be confirmed by a validated test (see sections 4.1 and 5.1). Posology The recommended dose of KEYTRUDA in adults is either 200 mg every 3 weeks or 400 mg every 6 weeks administered as an intravenous infusion over 30 minutes. The recommended dose of KEYTRUDA as monotherapy in paediatric patients aged 3 years and older with cHL or patients aged 12 years and older with melanoma is 2 mg/kg bodyweight (bw) (up to a maximum of 200 mg), every 3 weeks administered as an intravenous infusion over 30 minutes. For use in combination, see the Summary of Product Characteristics (SmPC) for the concomitant therapies. Patients should be treated with KEYTRUDA until disease progression or unacceptable toxicity (and up to maximum duration of therapy if specified for an indication). Atypical responses (i.e. an initial transient increase in tumour size or small new lesions within the first few months followed by tumour shrinkage) have been observed. It is recommended to continue treatment for clinically stable patients with initial evidence of disease progression until disease progression is confirmed. For the adjuvant treatment of melanoma, NSCLC, or RCC, KEYTRUDA should be administered until disease recurrence, unacceptable toxicity, or for a duration of up to one year. For the neoadjuvant and adjuvant treatment of TNBC, patients should be treated with neoadjuvant KEYTRUDA in combination with chemotherapy for 8 doses of 200 mg every 3 weeks or 4 doses of 400 mg every 6 weeks or until disease progression that precludes definitive surgery or unacceptable toxicity, followed by adjuvant treatment with KEYTRUDA as monotherapy for 9 doses of 200 mg every 3 weeks or 5 doses of 400 mg every 6 weeks or until disease recurrence or unacceptable toxicity. Patients who experience disease progression that precludes definitive surgery or unacceptable toxicity related to KEYTRUDA as neoadjuvant treatment in combination with chemotherapy should not receive KEYTRUDA monotherapy as adjuvant treatment. Dose delay or discontinuation (see also section 4.4) No dose reductions of KEYTRUDA are recommended. KEYTRUDA should be withheld or discontinued to manage adverse reactions as described in Table 1. Table 1: Recommended treatment modifications for KEYTRUDA Immune- mediated adverse reactions/Severity (Treatment modification) Pneumonitis: Grade 2 (Withhold until adverse reactions recover to Grades 0-1*), Grades 3 or 4, or recurrent Grade 2 (Permanently discontinue); Colitis: Grades 2 or 3 (Withhold until adverse reactions recover to Grades 0-1*), Grade 4 or recurrent Grade 3 (Permanently discontinue); Nephritis: Grade 2 with creatinine > 1.5 to ≤ 3 times upper limit of normal (ULN) (Withhold until adverse reactions recover to Grades 0-1*), Grade ≥ 3 with creatinine > 3 times ULN (Permanently discontinue); Endocrinopathies: Grade 2 adrenal insufficiency and hypophysitis ( Withhold treatment until controlled by hormone replacement), Grades 3 or 4 adrenal insufficiency or symptomatic hypophysitis, Type 1 diabetes associated with Grade ≥ 3 hyperglycaemia (glucose > 250 mg/dL or > 13.9 mmol/L) or associated with ketoacidosis, Hyperthyroidism Grade ≥ 3 (Withhold until adverse reactions recover to Grades 0-1* For patients with Grade 3 or Grade 4 endocrinopathies that improved to Grade 2 or lower and are controlled with hormone replacement, if indicated, continuation of pembrolizumab may be considered after corticosteroid taper, if needed. Otherwise treatment should be discontinued.), Hypothyroidism (Hypothyroidism may be managed with replacement therapy without treatment interruption.); Hepatitis: NOTE: for RCC patients treated with pembrolizumab in combination with axitinib with liver enzyme elevations, see dosing guidelines following this table. Grade 2 with aspartate aminotransferase (AST) or alanine aminotransferase (ALT) > 3 to 5 times ULN or total bilirubin > 1.5 to 3 times ULN (Withhold until adverse reactions recover to Grades 0-1*), Grade ≥ 3 with AST or ALT > 5 times ULN or total bilirubin > 3 times ULN (Permanently discontinue), In case of liver metastasis with baseline Grade 2 elevation of AST or ALT, hepatitis with AST or ALT increases ≥ 50% and lasts ≥ 1 week (Permanently discontinue); Skin reactions: Grade 3 or suspected Stevens-Johnson syndrome (SJS) or toxic epidermal necrolysis (TEN) (Withhold until adverse reactions recover to Grades 0-1*), Grade 4 or confirmed SJS or TEN (Permanently discontinue); Other immune-mediated adverse reactions: Based on severity and type of reaction (Grade 2 or Grade 3) (Withhold until adverse reactions recover to Grades 0-1*), Grades 3 or 4 myocarditis, Grades 3 or 4 encephalitis, Grades 3 or 4 Guillain-Barré syndrome (Permanently discontinue), Grade 4 or recurrent Grade 3 (Permanently discontinue). Infusion-related reactions: Grades 3 or 4 (Permanently discontinue). Note: toxicity grades are in accordance with National Cancer Institute Common Terminology Criteria for Adverse Events Version 4.0 (NCI-CTCAE v.4). * If treatment-related toxicity does not resolve to Grades 0-1 within 12 weeks after last dose of KEYTRUDA, or if corticosteroid dosing cannot be reduced to ≤ 10 mg prednisone or equivalent per day within 12 weeks, KEYTRUDA should be permanently discontinued. The safety of re-initiating pembrolizumab therapy in patients previously experiencing immune-mediated myocarditis is not known. KEYTRUDA, as monotherapy or as combination therapy, should be permanently discontinued for Grade 4 or recurrent Grade 3 immune-mediated adverse reactions, unless otherwise specified in Table 1. For Grade 4 haematological toxicity, only in patients with cHL, KEYTRUDA should be withheld until adverse reactions recover to Grades 0-1. KEYTRUDA in combination with axitinib in RCC For RCC patients treated with KEYTRUDA in combination with axitinib, see the SmPC regarding dosing of axitinib. When used in combination with pembrolizumab, dose escalation of axitinib above the initial 5 mg dose may be considered at intervals of six weeks or longer (see section 5.1). For liver enzyme elevations, in patients with RCC being treated with KEYTRUDA in combination with axitinib: • If ALT or AST ≥ 3 times ULN but < 10 times ULN without concurrent total bilirubin ≥ 2 times ULN, both KEYTRUDA and axitinib should be withheld until these adverse reactions recover to Grades 0-1. Corticosteroid therapy may be considered. Rechallenge with a single medicine or sequential rechallenge with both medicines after recovery may be considered. If rechallenging with axitinib, dose reduction as per the axitinib SmPC may be considered. • If ALT or AST ≥ 10 times ULN or > 3 times ULN with concurrent total bilirubin ≥ 2 times ULN, both KEYTRUDA and axitinib should be permanently discontinued and corticosteroid therapy may be considered. KEYTRUDA in combination with lenvatinib When used in combination with lenvatinib, one or both medicines should be interrupted as appropriate. Lenvatinib should be withheld, dose reduced, or discontinued in accordance with the instructions in the lenvatinib SmPC for combination with pembrolizumab. No dose reductions are recommended for KEYTRUDA. Patients treated with KEYTRUDA must be given the patient card and be informed about the risks of KEYTRUDA (see also package leaflet). Special populations Elderly No dose adjustment is necessary in patients ≥ 65 years (see sections 4.4 and 5.1). Renal impairment No dose adjustment is needed for patients with mild or moderate renal impairment. KEYTRUDA has not been studied in patients with severe renal impairment (see sections 4.4 and 5.2). Hepatic impairment No dose adjustment is needed for patients with mild or moderate hepatic impairment. KEYTRUDA has not been studied in patients with severe hepatic impairment (see sections 4.4 and 5.2). Paediatric population The safety and efficacy of KEYTRUDA in children below 18 years of age have not been established except in paediatric patients with melanoma or cHL. Currently available data are described in sections 4.8, 5.1 and 5.2. Method of administrationKEYTRUDA is for intravenous use. It must be administered by infusion over 30 minutes. KEYTRUDA must not be administered as an intravenous push or bolus injection. When administering KEYTRUDA as part of a combination with intravenous chemotherapy, KEYTRUDA should be administered first.For instructions on dilution of the medicinal product before administration, see section 6.6. 4.3 Contraindications Hypersensitivity to the active substance or to any of the excipients listed in section 6.1. 4.8 Undesirable effects Summary of the safety profile Pembrolizumab is most commonly associated with immune-mediated adverse reactions. Most of these, including severe reactions, resolved following initiation of appropriate medical therapy or withdrawal of pembrolizumab (see “Description of selected adverse reactions” below). The frequencies included below and in Table 2 are based on all reported adverse drug reactions, regardless of the investigator assessment of causality. Pembrolizumab in monotherapy (see section 4.2)The safety of pembrolizumab as monotherapy has been evaluated in 7,631 patients across tumour types and across four doses (2 mg/kg bw every 3 weeks, 200 mg every 3 weeks, or 10 mg/kg bw every 2 or 3 weeks) in clinical studies. In this patient population, the median observation time was 8.5 months (range: 1 day to 39 months) and the most frequent adverse reactions with pembrolizumab were fatigue (31%), diarrhoea (22%), and nausea (20%). The majority of adverse reactions reported for monotherapy were of Grades 1 or 2 severity. The most serious adverse reactions were immune-mediated adverse reactions and severe infusion-related reactions (see section 4.4). The incidences of immune‑mediated adverse reactions were 37% all Grades and 9% for Grades 3‑5 for pembrolizumab monotherapy in the adjuvant setting and 25% all Grades and 6% for Grades 3‑5 in the metastatic setting . No new immune‑mediated adverse reactions were identified in the adjuvant setting. Pembrolizumab in combination with chemotherapy (see section 4.2) When pembrolizumab is administered in combination, refer to the SmPC for the respective combination therapy components prior to initiation of treatment. The safety of pembrolizumab in combination with chemotherapy has been evaluated in 4,787 patients across tumour types receiving 200 mg, 2 mg/kg bw or 10 mg/kg bw pembrolizumab every 3 weeks, in clinical studies. In this patient population, the most frequent adverse reactions were anaemia (53%), nausea (51%), fatigue (35%), diarrhoea (34%), constipation (32%), vomiting (29%), decreased appetite (28%), neutropenia (28%), neutrophil count decreased (26%) and alopecia (25%). Incidences of Grades 3‑5 adverse reactions in patients with NSCLC were 67% for pembrolizumab combination therapy and 66% for chemotherapy alone, in patients with HNSCC were 85% for pembrolizumab combination therapy and 84% for chemotherapy plus cetuximab, in patients with oesophageal carcinoma were 86% for pembrolizumab combination therapy and 83% for chemotherapy alone, in patients with TNBC were 80% for pembrolizumab combination therapy and 77% for chemotherapy alone, in patients with cervical cancer were 82% for pembrolizumab combination and 75% for chemotherapy, with or without bevacizumab, in patients with gastric cancer were 75% for pembrolizumab combination therapy (chemotherapy with or without trastuzumab) and 79% for chemotherapy with or without trastuzumab, and in patients with biliary tract carcinoma were 85% for pembrolizumab combination therapy and 84% for chemotherapy alone. Pembrolizumab in combination with tyrosine kinase inhibitor (TKI) (see section 4.2) When pembrolizumab is administered in combination with axitinib or lenvatinib, refer to the SmPC for axitinib or lenvatinib prior to initiation of treatment. For additional lenvatinib safety information related to advanced RCC see the SmPC for Kisplyx and for advanced EC see the SmPC for Lenvima. For additional axitinib safety information for elevated liver enzymes see also section 4.4. The safety of pembrolizumab in combination with axitinib or lenvatinib in advanced RCC, and in combination with lenvatinib in advanced EC has been evaluated in a total of 1,456 patients with advanced RCC or advanced EC receiving 200 mg pembrolizumab every 3 weeks with either axitinib 5 mg twice daily or lenvatinib 20 mg once daily in clinical studies, as appropriate. In these patient populations, the most frequent adverse reactions were diarrhoea (58%), hypertension (54%), hypothyroidism (46%), fatigue (41%), decreased appetite (40%), nausea (40%), arthralgia (30%), vomiting (28%), weight decreased (28%), dysphonia (28%), abdominal pain (28%), proteinuria (27%), palmar plantar erythrodysaesthesia syndrome (26%), rash (26%), stomatitis (25%), constipation (25%), musculoskeletal pain (23%), headache (23%) and cough (21%). Grades 3 5 adverse reactions in patients with RCC were 80% for pembrolizumab in combination with either axitinib or lenvatinib and 71% for sunitinib alone. In patients with EC, Grades 3-5 adverse reactions were 89% for pembrolizumab in combination with lenvatinib and 73% for chemotherapy alone. Tabulated summary of adverse reactions Adverse reactions observed in clinical studies of pembrolizumab as monotherapy or in combination with chemotherapy or other anti-tumour medicines or reported from post-marketing use of pembrolizumab are listed in Table 2. These reactions are presented by system organ class and by frequency. Frequencies are defined as: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000); and not known (cannot be estimated from the available data). Within each frequency grouping, adverse reactions are presented in the order of decreasing seriousness. Adverse reactions known to occur with pembrolizumab or combination therapy components given alone may occur during treatment with these medicinal products in combination, even if these reactions were not reported in clinical studies with combination therapy. For additional safety information when pembrolizumab is administered in combination, refer to the SmPC for the respective combination therapy components. Table 2: Adverse reactions in patients treated with pembrolizumab†: Infections and infestationsMonotherapy:Common: pneumonia, In combination with chemotherapy:Common: pneumonia, In combination with axitinib or lenvatinib: Very common: urinary tract infection; common: pneumonia. Blood and lymphatic system disorders Monotherapy: Very Common: anaemia; Common: thrombocytopenia, neutropenia, lymphopenia; Uncommon: leukopenia, immune thrombocytopenia, eosinophilia; Rare: haemophagocytic lymphohistiocytosis, haemolytic anaemia, pure red cell aplasia, In combination with chemotherapy:Very Common:anaemia, neutropenia, thrombocytopenia; Common: febrile neutropenia, leukopenia, lymphopenia; Uncommon: eosinophilia; Rare: haemolytic anaemia, immune thrombocytopenia, in combination with axitinib or lenvatinib: Very common: anaemia, Common: neutropenia, thrombocytopenia, lymphopenia, leukopenia; Uncommon: eosinophilia. Immune system disorders Monotherapy:Common: infusion-related reaction⁎; Uncommon: sarcoidosis; Not known: solid organ transplant rejection, In combination with chemotherapy:Common: infusion-related reaction⁎; Rare: sarcoidosis, In combination with axitinib or lenvatinib: Common: infusion-related reaction⁎. Endocrine disorders Monotherapy: Very Common: hypothyroidism⁎; Common: hyperthyroidism; Uncommon: adrenal insufficiency⁎, hypophysitis⁎, thyroiditis⁎; Rare: hypoparathyroidism, In combination with chemotherapy:Very Common: hypothyroidism⁎, Common:, adrenal insufficiency⁎, thyroiditis⁎, hyperthyroidism⁎ Uncommon: hypophysitis⁎; Rare: hypoparathyroidism, In combination with axitinib or lenvatinib: Very common: hypothyroidism; Common: adrenal insufficiency⁎, hyperthyroidism, thyroiditis⁎; Uncommon: hypophysitis⁎; Rare: hypoparathyroidism, Metabolism and nutrition disorders Monotherapy: Very Common: decreased appetite; Common: hyponatraemia, hypokalaemia, hypocalcaemia; Uncommon: type 1 diabetes mellitus⁎, In combination with chemotherapy:Very common: hypokalaemia, decreased appetite; Common: hyponatraemia, hypocalcaemia; Uncommon: type 1 diabetes mellitus⁎, In combination with axitinib or lenvatinib Very common: decreased appetite; Common: hyponatraemia, hypokalaemia, hypocalcaemia; Uncommon: type 1 diabetes mellitus⁎. Psychiatric disorders Monotherapy: Common: insomnia, in combination with chemotherapy:Very Common: insomnia, in combination with axitinib or lenvatinib: Common: insomnia. Nervous system disorders Monotherapy: Very Common: headache; Common: dizziness, neuropathy peripheral, lethargy, dysgeusia; Uncommon: myasthenic syndrome⁎, epilepsy; Rare: Guillain-Barré syndrome⁎, encephalitis⁎, myelitis⁎, optic neuritis, meningitis (aseptic) ⁎, in combination with chemotherapy:Very common: neuropathy peripheral, headache; Common: dizziness, dysgeusia, lethargy; Uncommon: encephalitis⁎, epilepsy; Rare: myasthenic syndrome, Guillain-Barré Syndrome⁎, optic neuritis, in combination with axitinib or lenvatinib: Very common: headache; Common: dizziness, neuropathy peripheral, lethargy, dysgeusia; Uncommon: myasthenic syndrome⁎, encephalitis⁎; Rare: optic neuritis. Eye disorders Monotherapy: Common: dry eye; Uncommon: uveitis⁎; Rare: Vogt-Koyanagi-Harada syndrome, In combination with chemotherapy: Common: dry eye;Uncommon: uveitis⁎, In combination with axitinib or lenvatinib: Common: dry eye; Uncommon: uveitis⁎; Rare: Vogt-Koyanagi Harada-syndrome. Cardiac disorders Monotherapy: Common:cardiac arrhythmia‡ (including atrial fibrillation); Uncommon: myocarditis, pericardial effusion, pericarditis, In combination with chemotherapy: Common:cardiac arrhythmia‡ (including atrial fibrillation); Uncommon: myocarditis⁎, pericardial effusion, pericarditis, In combination with axitinib or lenvatinib: Common: cardiac arrhythmia‡ (including atrial fibrillation); Uncommon: myocarditis; pericardial effusion. Vascular disorders Monotherapy: Common: hypertension; Rare: vasculitis⁎, In combination with chemotherapy: Common: hypertension; Uncommon: vasculitis⁎, In combination with axitinib or lenvatinib: Very common: hypertension; Uncommon: vasculitis⁎. Respiratory, thoracic and mediastinal disorders Monotherapy: Very Common: dyspnoea, cough; Common: pneumonitis⁎, In combination with chemotherapy:Very common: dyspnoea, cough;Common: pneumonitis⁎, In combination with axitinib or lenvatinib: Very common: dyspnoea, cough; Common: pneumonitis⁎. Gastrointestinal disorders Monotherapy: Very common: diarrhoea, abdominal pain⁎, nausea, vomiting, constipation; Common: colitis⁎, dry mouth; Uncommon: pancreatitis⁎, gastritis⁎, gastrointestinal ulceration⁎; Rare: small intestinal perforation, In combination with chemotherapy: Very common: diarrhoea, vomiting, nausea, abdominal pain⁎, constipation; Common: colitis⁎, gastritis⁎, dry mouth; Uncommon: pancreatitis⁎, gastrointestinal ulceration⁎; Rare: small intestinal perforation; In combination with axitinib or lenvatinib: Very Common: diarrhoea, abdominal pain⁎, nausea, vomiting, constipation; Common: colitis⁎, pancreatitis⁎, gastritis⁎, dry mouth;Uncommon: gastrointestinal ulceration⁎; Rare: small intestinal perforation. Hepatobiliary disorders Monotherapy: Common: hepatitis⁎; Rare: cholangitis sclerosing, In combination with chemotherapy: Common: hepatitis⁎, Rare: cholangitis sclerosing⁎, In combination with axitinib or lenvatinib: Common: hepatitis⁎. Skin and subcutaneous tissue disorders Monotherapy: Very common: pruritus⁎, rash⁎; Common: severe skin reactions⁎, erythema, dermatitis, dry skin, vitiligo⁎, eczema, alopecia, dermatitis acneiform; Uncommon: psoriasis, lichenoid keratosis⁎, papule, hair colour changes; Rare: Stevens-Johnson syndrome, erythema nodosum, toxic epidermal necrolysis, In combination with chemotherapy: Very common: alopecia, pruritus⁎, rash⁎; Common: severe skin reactions⁎, erythema, dermatitis, dry skin, dermatitis acneiform, eczema; Uncommon: psoriasis, lichenoid keratosis⁎, vitiligo⁎, papule; Rare: Stevens‑Johnson syndrome, erythema nodosum, hair colour changes, In combination with axitinib or lenvatinib: Very common: rash⁎, pruritus⁎; Common: severe skin reactions⁎, dermatitis, dry skin, erythema, dermatitis acneiform, alopecia; Uncommon: eczema, lichenoid keratosis⁎, psoriasis, vitiligo⁎, papule, hair colour changes; Rare: toxic epidermal necrolysis, Stevens Johnson syndrome. Musculoskeletal and connective tissue disorders Monotherapy: Very Common: musculoskeletal pain⁎, arthralgia; Common: myositis⁎, pain in extremity, arthritis⁎ Uncommon: tenosynovitis⁎; Rare: Sjogren’s syndrome, In combination with chemotherapy: Very Common: musculoskeletal pain⁎, arthralgia ;Common: myositis⁎, pain in extremity, arthritis⁎; Uncommon: tenosynovitis⁎; Rare: Sjogren’s syndrome, In combination with axitinib or lenvatinib: Very common: arthralgia, musculoskeletal pain⁎, myositis⁎, pain in extremity; Common: arthritis⁎; Uncommon: tenosynovitis⁎; Rare: Sjogren’s syndrome. Renal and urinary disorders Monotherapy: Uncommon: nephritis⁎; Rare: cystitis noninfective, in combination with chemotherapy: Common: acute kidney injury; Uncommon: nephritis⁎, cystitis noninfective, in combination with axitinib or lenvatinib: Common: nephritis⁎; Rare: cystitis noninfective. General disorders and administration site conditions Monotherapy: Very common: fatigue, asthenia, oedema⁎, pyrexia; Common: influenza like illness, chills, In combination with chemotherapy: Very common: fatigue, asthenia, pyrexia ⁎; Common: oedema, influenza like illness, chills, In combination with axitinib or lenvatinib: Very common: fatigue, asthenia, oedema⁎, pyrexia, Common: influenza like illness, chills. Investigations Monotherapy: Common: alanine aminotransferase increased, aspartate aminotransferase increased, blood alkaline phosphatase increased, hypercalcaemia, blood bilirubin increased, blood creatinine increased; Uncommon: amylase increased, In combination with chemotherapy:Very common: alanine aminotransferase increased, aspartate aminotransferase increased; Common: blood bilirubin increased, blood alkaline phosphatase increased, blood creatinine increased, hypercalcaemia; Uncommon: amylase increased, In combination with axitinib or lenvatinib: Very common: lipase increased, alanine aminotransferase increased, aspartate aminotransferase increased, blood creatinine increased; Common: amylase increased, blood bilirubin increased, blood alkaline phosphatase increased, hypercalcaemia. † Adverse reaction frequencies presented in Table 2 may not be fully attributable to pembrolizumab alone but may contain contributions from the underlying disease or from other medicinal products used in a combination. ‡Based upon a standard query including bradyarrhythmias and tachyarrhythmias. ⁎The following terms represent a group of related events that describe a medical condition rather than a single event: infusion-related reaction (drug hypersensitivity, anaphylactic reaction, anaphylactoid reaction, hypersensitivity, infusion related hypersensitivity reaction, cytokine release syndrome and serum sickness); sarcoidosis (cutaneous sarcoidosis and pulmonary sarcoidosis); hypothyroidism (myxoedema, immune-mediated hypothyroidism and autoimmune hypothyroidism); adrenal insufficiency (Addison’s disease, adrenocortical insufficiency acute and secondary adrenocortical insufficiency); thyroiditis (autoimmune thyroiditis, silent thyroiditis, thyroid disorder, thyroiditis acute, and immune-mediated thyroiditis); hyperthyroidism (Basedow’s disease); hypophysitis (hypopituitarism andlymphocytic hypophysitis); type 1 diabetes mellitus (diabetic ketoacidosis); myasthenic syndrome (myasthenia gravis, including exacerbation); encephalitis (autoimmune encephalitis and noninfective encephalitis); Guillain-Barré syndrome (axonal neuropathy and demyelinating polyneuropathy); myelitis (including transverse myelitis); meningitis aseptic (meningitis and meningitis noninfective); uveitis (chorioretinitis, iritis and iridocyclitis); n. myocarditis (autoimmune myocarditis); vasculitis (central nervous system vasculitis, aortitis and giant cell arteritis); pneumonitis (interstitial lung disease, organising pneumonia, immune-mediated pneumonitis, immune-mediated lung disease and autoimmune lung disease); abdominal pain (abdominal discomfort, abdominal pain upper and abdominal pain lower); colitis (colitis microscopic, enterocolitis, enterocolitis haemorrhagic, autoimmune colitis, and immune-mediated enterocolitis); gastritis (gastritis erosive and gastritis haemorrhagic); pancreatitis (autoimmune pancreatitis, pancreatitis acute and immune-mediated pancreatitis); gastrointestinal ulceration (gastric ulcer and duodenal ulcer); hepatitis (autoimmune hepatitis, immune-mediated hepatitis, drug induced liver injury and acute hepatitis); cholangitis sclerosing (immune-mediated cholangitis); pruritus (urticaria, urticaria papular and pruritus genital); rash (rash erythematous, rash macular, rash maculo‑papular, rash papular, rash pruritic, rash vesicular and genital rash); severe skin reactions (exfoliative rash, pemphigus, and Grade ≥ 3 of the following: cutaneous vasculitis, dermatitis bullous, dermatitis exfoliative, dermatitis exfoliative generalised, erythema multiforme, lichen planus, oral lichen planus, pemphigoid, pruritus, pruritus genital, rash, rash erythematous, rash maculo‑papular, rash pruritic, rash pustular, skin necrosis and toxic skin eruption); vitiligo (skin depigmentation, skin hypopigmentation and hypopigmentation of the eyelid); lichenoid keratosis (lichen planus and lichen sclerosus); musculoskeletal pain (musculoskeletal discomfort, back pain, musculoskeletal stiffness, musculoskeletal chest pain and torticollis); cc. myositis (myalgia, myopathy, necrotising myositis, polymyalgia rheumatica and rhabdomyolysis); arthritis (joint swelling, polyarthritis, joint effusion, autoimmune arthritis and immune-mediated arthritis); tenosynovitis (tendonitis, synovitis and tendon pain); nephritis (autoimmune nephritis, immune-mediated nephritis, tubulointerstitial nephritis and renal failure, renal failure acute, or acute kidney injury with evidence of nephritis, nephrotic syndrome, glomerulonephritis, glomerulonephritis membranous and glomerulonephritis acute); oedema (oedema peripheral, generalised oedema, fluid overload, fluid retention, eyelid oedema and lip oedema, face oedema, localised oedema and periorbital oedema). Description of selected adverse reactions Data for the following immune‑mediated adverse reactions are based on patients who received pembrolizumab across four doses (2 mg/kg bw every 3 weeks, 10 mg/kg bw every 2 or 3 weeks, or 200 mg every 3 weeks) in clinical studies (see section 5.1). The management guidelines for these adverse reactions are described in section 4.4. Immune‑mediated adverse reactions (see section 4.4) Immune‑mediated pneumonitis Pneumonitis occurred in 324 (4.2%) patients, including Grade 2, 3, 4 or 5 cases in 143 (1.9%), 81 (1.1%), 19 (0.2%) and 9 (0.1%) patients, respectively, receiving pembrolizumab. The median time to onset of pneumonitis was 3.9 months (range: 2 days to 27.2 months). The median duration was 2.0 months (range: 1 day to 51.0+ months). Pneumonitis occurred more frequently in patients with a history of prior thoracic radiation (8.1%) than in patients who did not receive prior thoracic radiation (3.9%). Pneumonitis led to discontinuation of pembrolizumab in 131 (1.7%) patients. Pneumonitis resolved in 196 patients, 6 with sequelae. In patients with NSCLC, pneumonitis occurred in 206 (6.1%), including Grade 2, 3, 4 or 5 cases in 92 (2.7%), 56 (1.7%), 16 (0.5%) and 9 (0.43%), respectively. In patients with locally advanced or metastatic NSCLC, pneumonitis occurred in 8.9% with a history of prior thoracic radiation. In patients with cHL, the incidence of pneumonitis (all Grades) ranged from 5.2% to 10.8% for cHL patients in KEYNOTE-087 (n=210) and KEYNOTE-204 (n=148), respectively. Immune‑mediated colitis Colitis occurred in 158 (2.1%) patients, including Grade 2, 3 or 4 cases in 49 (0.6%), 82 (1.1%) and 6 (0.1%) patients, respectively, receiving pembrolizumab. The median time to onset of colitis was 4.3 months (range: 2 days to 24.3 months). The median duration was 1.1 month (range: 1 day to 45.2 months). Colitis led to discontinuation of pembrolizumab in 48 (0.6%) patients. Colitis resolved in 132 patients, 2 with sequelae. In patients with CRC treated with pembrolizumab as monotherapy (n=153), the incidence of colitis was 6.5% (all Grades) with 2.0% Grade 3 and 1.3% Grade 4. Immune-mediated hepatitisHepatitis occurred in 80 (1.0%) patients, including Grade 2, 3 or 4 cases in 12 (0.2%), 55 (0.7%) and 8 (0.1%) patients, respectively, receiving pembrolizumab. The median time to onset of hepatitis was 3.5 months (range: 8 days to 26.3 months). The median duration was 1.3 months (range: 1 day to 29.0+ months). Hepatitis led to discontinuation of pembrolizumab in 37 (0.5%) patients. Hepatitis resolved in 60 patients.Immune-mediated nephritis Nephritis occurred in 37 (0.5%) patients, including Grade 2, 3 or 4 cases in 11 (0.1%), 19 (0.2%) and 2 (< 0.1%) patients, respectively, receiving pembrolizumab as monotherapy. The median time to onset of nephritis was 4.2 months (range: 12 days to 21.4 months). The median duration was 3.3 months (range: 6 days to 28.2+ months). Nephritis led to discontinuation of pembrolizumab in 17 (0.2%) patients. Nephritis resolved in 25 patients, 5 with sequelae. In patients with non-squamous NSCLC treated with pembrolizumab in combination with pemetrexed and platinum chemotherapy (n=488), the incidence of nephritis was 1.4% (all Grades) with 0.8% Grade 3 and 0.4% Grade 4.Immune-mediated endocrinopathiesAdrenal insufficiency occurred in 74 (1.0%) patients, including Grade 2, 3 or 4 cases in 34 (0.4%), 31 (0.4%) and 4 (0.1%) patients, respectively, receiving pembrolizumab. The median time to onset of adrenal insufficiency was 5.4 months (range: 1 day to 23.7 months). The median duration was not reached (range: 3 days to 40.1+ months). Adrenal insufficiency led to discontinuation of pembrolizumab in 13 (0.2%) patients. Adrenal insufficiency resolved in 28 patients, 11 with sequelae. Hypophysitis occurred in 52 (0.7%) patients, including Grade 2, 3 or 4 cases in 23 (0.3%), 24 (0.3%) and 1 (< 0.1%) patients, respectively, receiving pembrolizumab. The median time to onset of hypophysitis was 5.9 months (range: 1 day to 17.7 months). The median duration was 3.6 months (range: 3 days to 48.1+ months). Hypophysitis led to discontinuation of pembrolizumab in 14 (0.2%) patients. Hypophysitis resolved in 23 patients, 8 with sequelae. Hyperthyroidism occurred in 394 (5.2%) patients, including Grade 2 or 3 cases in 108 (1.4%) and 9 (0.1%) patients, respectively, receiving pembrolizumab. The median time to onset of hyperthyroidism was 1.4 months (range: 1 day to 23.2 months). The median duration was 1.6 months (range: 4 days to 43.1+ months). Hyperthyroidism led to discontinuation of pembrolizumab in 4 (0.1%) patients. Hyperthyroidism resolved in 326 (82.7%) patients, 11 with sequelae. In patients with melanoma, NSCLC and RCC treated with pembrolizumab monotherapy in the adjuvant setting (n=2,060), the incidence of hyperthyroidism was 11.0%, the majority of which were Grade 1 or 2. Hypothyroidism occurred in 939 (12.3%) patients, including Grade 2 or 3 cases in 687 (9.0%) and 8 (0.1%) patients, respectively, receiving pembrolizumab. The median time to onset of hypothyroidism was 3.4 months (range: 1 day to 25.9 months). The median duration was not reached (range: 2 days to 63.0+ months). Hypothyroidism led to discontinuation of pembrolizumab in 6 (0.1%) patients. Hypothyroidism resolved in 216 (23.0%) patients, 16 with sequelae. In patients with cHL (n=389) the incidence of hypothyroidism was 17%, all of which were Grade 1 or 2. In patients with HNSCC treated with pembrolizumab as monotherapy (n=909), the incidence of hypothyroidism was 16.1% (all Grades) with 0.3% Grade 3. In patients with HNSCC treated with pembrolizumab in combination with platinum and 5-FU chemotherapy (n=276), the incidence of hypothyroidism was 15.2%, all of which were Grade 1 or 2. In patients treated with pembrolizumab in combination with axitinib or lenvatinib (n=1,456), the incidence of hypothyroidism was 46.2% (all Grades) with 0.8% Grade 3 or 4. In patients with melanoma, NSCLC and RCC treated with pembrolizumab monotherapy in the adjuvant setting (n=2,060), the incidence of hypothyroidism was 18.5%, the majority of which were Grade 1 or 2. Immune-mediated skin adverse reactions Immune‑mediated severe skin reactions occurred in 130 (1.7%) patients, including Grade 2, 3, 4 or 5 cases in 11 (0.1%), 103 (1.3%), 1 (< 0.1%) and 1 (< 0.1%) patients, respectively, receiving pembrolizumab. The median time to onset of severe skin reactions was 2.8 months (range: 2 days to 25.5 months). The median duration was 1.9 months (range: 1 day to 47.1+ months). Severe skin reactions led to discontinuation of pembrolizumab in 18 (0.2%) patients. Severe skin reactions resolved in 95 patients, 2 with sequelae. Rare cases of SJS and TEN, some of them with fatal outcome, have been observed (see sections 4.2 and 4.4). Complications of allogeneic HSCT in cHL Of 14 patients in KEYNOTE‑013 who proceeded to allogeneic HSCT after treatment with pembrolizumab, 6 patients reported acute GVHD and 1 patient reported chronic GVHD, none of which were fatal. Two patients experienced hepatic VOD, one of which was fatal. One patient experienced engraftment syndrome post-transplant. Of 32 patients in KEYNOTE‑087 who proceeded to allogeneic HSCT after treatment with pembrolizumab, 16 patients reported acute GVHD and 7 patients reported chronic GVHD, two of which were fatal. No patients experienced hepatic VOD. No patients experienced engraftment syndrome post-transplant. Of 14 patients in KEYNOTE‑204 who proceeded to allogeneic HSCT after treatment with pembrolizumab, 8 patients reported acute GVHD and 3 patients reported chronic GVHD, none of which were fatal. No patients experienced hepatic VOD. One patient experienced engraftment syndrome post-transplant. Elevated liver enzymes when pembrolizumab is combined with axitinib in RCC In a clinical study of previously untreated patients with RCC receiving pembrolizumab in combination with axitinib, a higher than expected incidence of Grades 3 and 4 ALT increased (20%) and AST increased (13%) were observed. The median time to onset of ALT increased was 2.3 months (range: 7 days to 19.8 months). In patients with ALT ≥ 3 times ULN (Grades 2-4, n=116), ALT resolved to Grades 0-1 in 94%. Fifty-nine percent of the patients with increased ALT received systemic corticosteroids. Of the patients who recovered, 92 (84%) were rechallenged with either pembrolizumab (3%) or axitinib (31%) monotherapy or with both (50%). Of these patients, 55% had no recurrence of ALT > 3 times ULN, and of those patients with recurrence of ALT > 3 times ULN, all recovered. There were no Grade 5 hepatic events. Laboratory abnormalities In patients treated with pembrolizumab monotherapy, the proportion of patients who experienced a shift from baseline to a Grade 3 or 4 laboratory abnormality was as follows:9.4% for lymphocytes decreased, 7.4% for sodium decreased, 5.8% for haemoglobin decreased, 5.3% for phosphate decreased, 5.3% for glucose increased, 3.3% for ALT increased, 3.1% for AST increased, 2.6% for alkaline phosphatase increased, 2.3% for potassium decreased, 2.1% for potassium increased, 1.9% for neutrophils decreased, 1.8% for platelets decreased, 1.8% for calcium increased, 1.7% for bilirubin increased, 1.5% for calcium decreased, 1.4% for albumin decreased, 1.3% for creatinine increased, 1.2% for glucose decreased, 0.8% for leucocytes decreased, 0.7% for magnesium increased, 0.5% for sodium increased, 0.4% for haemoglobin increased, and 0.2% for magnesium decreased. In patients treated with pembrolizumab in combination with chemotherapy, the proportion of patients who experienced a shift from baseline to a Grade 3 or 4 laboratory abnormality was as follows: 41.3% for neutrophils decreased, 26.9% for lymphocytes decreased, 24.8% for leucocytes decreased, 21.9% for haemoglobin decreased, 14.9% for platelets decreased, 11.0% for sodium decreased, 8.7% for potassium decreased, 8.1% for phosphate decreased, 6.1% for ALT increased, 5.7% for AST increased, 5.6% for glucose increased, 4.1% for bilirubin increased, 3.8% for calcium decreased, 3.5% for creatinine increased, 3.4% for potassium increased, 3.2% for alkaline phosphatase increased, 2.9% for albumin decreased, 1.8% for calcium increased, 1.1% for glucose decreased, 0.5% for sodium increased and 0.1% for haemoglobin increased. In patients treated with pembrolizumab in combination with axitinib or lenvatinib, the proportion of patients who experienced a shift from baseline to a Grade 3 or 4 laboratory abnormality was as follows: 23.0% for lipase increased (not measured in patients treated with pembrolizumab and axitinib), 12.0% for lymphocyte decreased, 11.4% for sodium decreased, 11.2% for amylase increased, 11.2% for triglycerides increased, 10.4% for ALT increased, 8.9% for AST increased, 7.8% for glucose increased, 6.8% for phosphate decreased, 6.1% for potassium decreased, 5.1% for potassium increased, 4.5% for cholesterol increased, 4.4% for creatinine increased, 4.2% for haemoglobin decreased, 4.0% for magnesium decreased, 3.5% for neutrophils decreased, 3.1% for alkaline phosphatase increased, 3.0% for platelets decreased, 2.8% for bilirubin increased, 2.2% for calcium decreased, 1.7% for white blood cells decreased, 1.6% for magnesium increased, 1.5% for prothrombin INR increased, 1.4% for glucose decreased, 1.2% for albumin decreased, 1.2% for calcium increased, 0.4% for sodium increased, and 0.1% for haemoglobin increased. Immunogenicity In clinical studies in patients treated with pembrolizumab 2 mg/kg bw every three weeks, 200 mg every three weeks, or 10 mg/kg bw every two or three weeks as monotherapy, 36 (1.8%) of 2,034 evaluable patients tested positive for treatment‑emergent antibodies to pembrolizumab, of which 9 (0.4%) patients had neutralising antibodies against pembrolizumab. There was no evidence of an altered pharmacokinetic or safety profile with anti-pembrolizumab binding or neutralising antibody development. Paediatric population The safety of pembrolizumab as monotherapy has been evaluated in 161 paediatric patients aged 9 months to 17 years with advanced melanoma, lymphoma, or PD-L1 positive advanced, relapsed, or refractory solid tumours at 2 mg/kg bw every 3 weeks in the Phase I/II study KEYNOTE-051. The cHL population (n=22) included patients 11 to 17 years of age. The safety profile in paediatric patients was generally similar to that seen in adults treated with pembrolizumab. The most common adverse reactions (reported in at least 20% of paediatric patients) were pyrexia (33%), vomiting (30%), headache (26%), abdominal pain (22%), anaemia (21%), cough (21%) and constipation (20%). The majority of adverse reactions reported for monotherapy were of Grades 1 or 2 severity. Seventy-six (47.2%) patients had 1 or more Grades 3 to 5 adverse reactions of which 5 (3.1%) patients had 1 or more adverse reactions that resulted in death. The frequencies are based on all reported adverse drug reactions, regardless of the investigator assessment of causality. Long-term safety data of pembrolizumab in adolescents with Stage IIB, IIC and III melanoma treated in the adjuvant setting are currently unavailable. Reporting of suspected adverse reactions Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system: in Belgium: Agence Fédérale des Médicaments et des Produits de Santé. Division Vigilance. Boîte Postale 97, B-1000 Brussels Madou. Website: www.notifieruneffetindesirable.be, e-mail: adr@afmps.be, in Luxembourg: Centre Régional de Pharmacovigilance de Nancy ou Division de la pharmacie et des médicaments de la Direction de la santé. Site internet: www.guichet.lu/pharmacovigilance. 7. MARKETING AUTHORISATION HOLDER Merck Sharp & Dohme B.V. Waarderweg 39. 2031 BN Haarlem. The Netherlands. 8. MARKETING AUTHORISATION NUMBER(S) EU/1/15/1024/002 9. DATE OF FIRST AUTHORISATION/RENEWAL OF THE AUTHORISATION Date of first authorisation: 17 July 2015. Date of latest renewal: 24 March 2020. 10. DATE OF REVISION OF THE TEXT 12/2023. Detailed information on this medicinal product is available on the website of the European Medicines Agency http://www.ema.europa.eu. DELIVERY: on medical prescription.